

 下载:
下载:

-
滇川干热河谷地处我国西南生物多样性中心横断山脉的南缘,主要包括元江、怒江、金沙江和澜沧江四大干热河谷区,因地理位置、地形地貌及气候等因素,表现出独具特色的干、热生态特点[1]。由于气候和人类活动等因素的影响,干热河谷区域植被的原生类型几乎不复存在,现存的主要是与非洲萨王纳植被(即热带草原植被)具有一定相似性的半稀树草原植被[1],有的地方已变成光秃裸露的荒山,生物多样性和生态系统的稳定性遭到破坏,水土流失严重。生长在该地区的植物类群对这种严酷的干热生境形成了很好的适应性[2], 余甘子就是其中典型的代表。
余甘子(Phyllanthus emblica L.)隶属叶下珠科(Phyllanthaceae)叶下珠属(Phyllanthus),是一种分布于热带和亚热带地区的重要食药同源经济树种,被世界卫生组织列为在世界范围内推广种植的三种保健植物之一[3]。其叶片、根和果实均含有维生素C、类黄酮、超氧化物歧化酶等有益于健康的成分,多种治疗功效在现代医药学研究中已被证实[4-5]。其中,余甘子果实是维生素C最丰富的天然来源之一,具有极高的商业开发潜力[6]。在生态学方面,余甘子因极耐干旱贫瘠环境而常被作为我国西南干热河谷荒山绿化的先锋树种,对水土流失严重的干热河谷地带有明显的保水、固土作用[3]。尽管兼具药用、经济和生态价值,但目前余甘子遗传背景仍然不清楚,特别是该植物的分子生物学背景研究较为薄弱。
随着高通量测序技术的发展,测序时间和成本显著减少。对于无参考基因组的物种,采用转录组高通量测序技术可获取大量的数据信息,构建cDNA文库,挖掘重要功能基因,同时也为大量分子标记的开发奠定基础,是开展植物优良性状研究的重要手段[7]。鉴于此,本研究以云南宾川干热河谷地区的余甘子为研究材料,采用Illumina Hiseq 4000平台对余甘子进行高通量转录组测序,并对测序的原始数据进行过滤和de novo组装,之后通过生物信息学的方法对获得的Unigenes进行功能注释、CDS预测、TF及抗性基因预测等分析,以期了解余甘子在一定生长发育时期基因表达情况及功能分布特征,为余甘子转录组水平上的研究以及微卫星标记的开发和抗旱相关基因的挖掘奠定基础,亦为干热河谷地区的生态恢复和经济发展提供参考。
-
试验材料采自云南省宾川县(25°45′59″ N, 100°26′29″ E,属金沙江干热河谷区),选择5株相距15 m以上的健康余甘子植株,每株各采集嫩叶5 g左右分别放在锡箔纸中包好,迅速放入液氮中,然后保存于-80℃冰箱中备用。
-
各单株分别称取0.1 g叶片等量混合均匀后,按照Kumar等[8]的方法提取总RNA。RNA样品的浓度、纯度和完整性分别用Qubit 2.0、Nanodrop和Agilent 2100检测。以片段化的mRNA为模板,合成双链cDNA,纯化后进行末端修复、加A尾和接头,经片段大小筛选后进行PCR扩增富集得到cDNA文库。构建好的文库用Agilent 2100 Bioanalyzer质检合格后,使用Illumina HiSeq 4000进行测序。
-
为确保所获得reads的精确性,首先对原始数据进行一系列过滤(去除包含接头的reads、未知核苷酸比例>10%的reads以及质量值Q < 10的碱基数占整个read 40%以上的reads),过滤后得到Clean reads。然后使用Trinity软件[9]对过滤后的Clean reads进行de novo组装获得转录本。最后使用CD-HIT[10]对转录本进行聚类得到Unigenes。
-
通过BLASTx将获得的Unigenes序列比对到NR、COG、KEGG以及SwissProt四大蛋白数据库,获得Unigenes序列的蛋白功能注释信息(E-value≤1e-5)。通过BLASTp将Unigenes比对到PlantTFDB和R-Gene数据库(PRGdb)分别预测TF家族和抗性基因。
-
测序共产生10.95 Gb的Raw reads,经过滤处理后获得10.52 Gb的Clean reads,占原始读长的98.03%。其中,中间未知碱基(N)含量为0,Q20为98.47%,Q30为95.28%,GC含量为43.29%。说明余甘子转录组测序获得序列的数量、质量和精确性均较高,为下一步de novo组装提供了较好的原始序列数据。
通过组装共获得97 628条转录本,平均长度、GC含量分别为648 nt和39.69%,N50长度较长,为1 116 nt,表明组装质量较好。进一步聚类去冗余得到76 881条Unigenes,平均长度、GC含量及N50长度分别为713 nt、39.74%和1 257 nt。对组装的转录本长度进一步分析可知(图 1A):22.34%的转录本长度在1 000 nt以上,仅1.79%的转录本≥3 000 nt。对聚类去冗余后Unigenes的长度分布进行统计(图 1B),有23.57%的序列长度大于1 000 nt,2.10%的转录本序列大于3 000 nt。由此可以看出:经Trinity软件组装后Unigenes的平均长度及N50长度有所增加,1 000 nt及以上序列所占比例有所提高,表明组装及聚类去冗余效果较好,可进行后续分析。
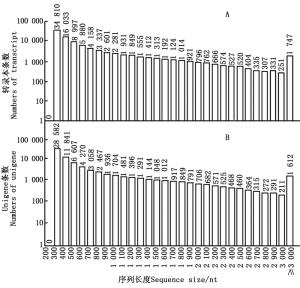
图 1 余甘子转录本和Unigenes的长度分布图
Figure 1. Transcripts and Unigenes length distribution for P. emblica
-
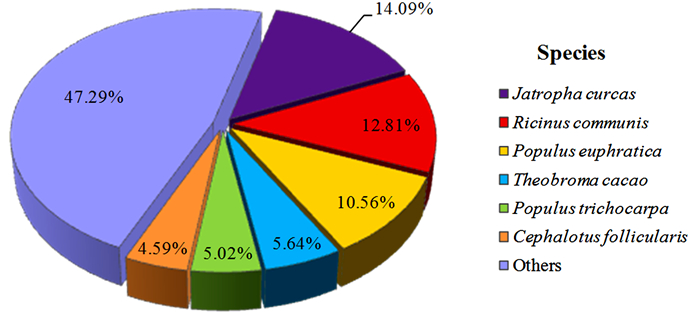
将76 881条Unigenes比对到NR等四大蛋白数据库(E-value≤1e-5),共有44 768条Unigenes获得注释,注释率为58.23%。其中,在NR数据库中获得注释的Unigenes数量最多,为44 270条(57.58%),且与其它物种同源序列具有不同程度的匹配度。由图 2可见:注释序列分布较多的3个物种分别是麻疯树(Jatropha curcas L.)、蓖麻(Ricinus communis L.)、胡杨(Populus euphratica Oliv.),分别占NR数据库所注释Unigenes总数的14.09%、12.81%、10.56%,其余近一半的被注释Unigenes分布于其它542个物种中。由于缺乏余甘子基因组和转录组信息,仍有一部分Unigenes未能在NR库中匹配到同源序列。

图 2 余甘子转录组Unigenes的NR注释物种分布
Figure 2. NR annotated species distribution of Unigenes of transcriptome in P. emblica
-
COG数据库是识别直系同源基因并对基因产物进行同源分类的数据库。共有27 008条Unigenes(35.13%)分别被注释到25个COG功能分类中,共包含42 611个功能注释信息,基本涉及到余甘子大部分的生命活动(图 3)。各类功能的基因表达丰度各不相同,以一般功能基因为最大类别,占COG数据库总功能注释信息的19.77%;其次是信号转导机制(11.58%)与翻译后修饰、蛋白质折叠和分子伴侣(11.24%);而细胞能动性基因较少,仅有22(0.05%)个。
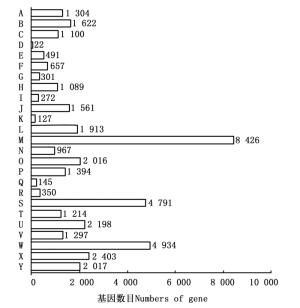
图 3 余甘子转录组Unigenes的COG功能注释分布统计图
Figure 3. COG functional annotation distribution of Unigenes of transcriptome in P. emblica
-
根据NR注释信息对余甘子所有Unigenes进行GO功能分类统计,从宏观上认识该物种叶片中相关基因功能分布特征。分析图 4可知:有19 749(25.69%)条Unigenes得到97 102个GO注释信息,平均每条4.9个;近3/4 Unigenes在GO库中未匹配到同源序列,进一步说明余甘子基因信息不足。
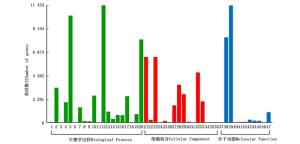
图 4 余甘子转录组Unigenes的GO功能分类统计图
Figure 4. GO functional classification of Unigenes of transcriptome in P. emblica
获得的GO注释可划分为生物学过程、细胞组分和分子功能三大类,其中执行生物学过程的注释最多(占47.91%),涉及分子功能的最少(占22.45%)。这三大功能分类又被细分为47个功能亚类。参与生物学过程的46 519个GO条目被划分为20个亚类,其中,以代谢过程最多,占该类型的24.62%。构成细胞组分的28 786个GO条目被划分为16个亚类,以细胞、细胞部分以及细胞器较多,分别占该类型的22.32%、22.31%和17.06%。分子功能包含的11个亚类中,以催化活性和结合居多,分别占该类别的52.48%和38.23%。总体来说,余甘子在细胞活动、代谢活动的基因表达丰度较高,表明余甘子自身具有较强的代谢能力。
-
KEGG是系统分析基因产物在细胞中的代谢途径以及这些基因产物功能的数据库,利用KEGG可以进一步研究基因在生物学上的复杂行为。余甘子KEGG比对结果表明,获得注释的18 175(23.64%)条Unigenes参与的代谢通路可归为6大类别、21个亚类。6大类代谢通路中,代谢和遗传信息处理相关的通路所占比例最大,分别为58.42%和26.66%;比例最小的是人类疾病相关的通路,仅占1.26%;细胞过程、环境信息处理以及生物系统相关的通路分别占5.65%、4.21%和3.82%。进一步细分为21个亚类,其中,代谢包含的11个亚类中,碳水化合物代谢所占比例最高,为13.79%,而糖类生物合成与代谢所占比例最低,为1.71%。遗传信息处理包含4个亚类,以翻译(10.89%)与折叠、分类和降解(8.41%)为主要代谢途径,而复制和修复(3.71%)以及转录(3.65%)在此时期代谢较弱。总的来说,该时期余甘子代谢活动和遗传信息处理能力较强。
-
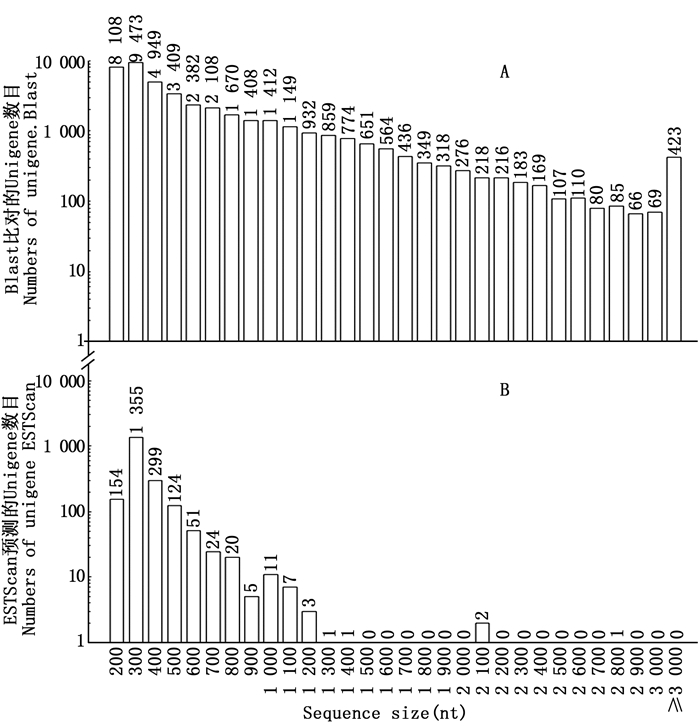
对CDS进行预测能为余甘子功能基因的挖掘利用和分子标记的开发提供重要参考。按照NR、SwissProt、KEGG和COG数据库的优先级顺序,通过BLAST比对共获得42 953条Unigenes的CDS(E≤1e-5),与以上蛋白数据库皆比对不上的Unigenes用软件ESTScan[11]预测后获得2 058个CDS。从图 5A可看出:通过BLAST获得的CDS序列长度较长,≥1 000 nt的占21.99%;而通过ESTScan预测后获得的CDS序列较短,主要集中在200600 nt(占96.36%)(图 5B)。
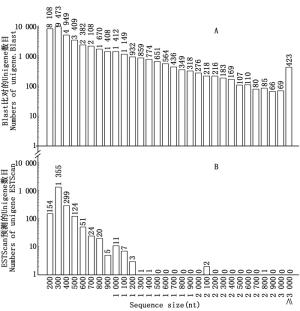
图 5 余甘子转录组Unigenes的CDS长度分布图
Figure 5. CDS length distribution of Unigenes of transcriptome in P. emblica
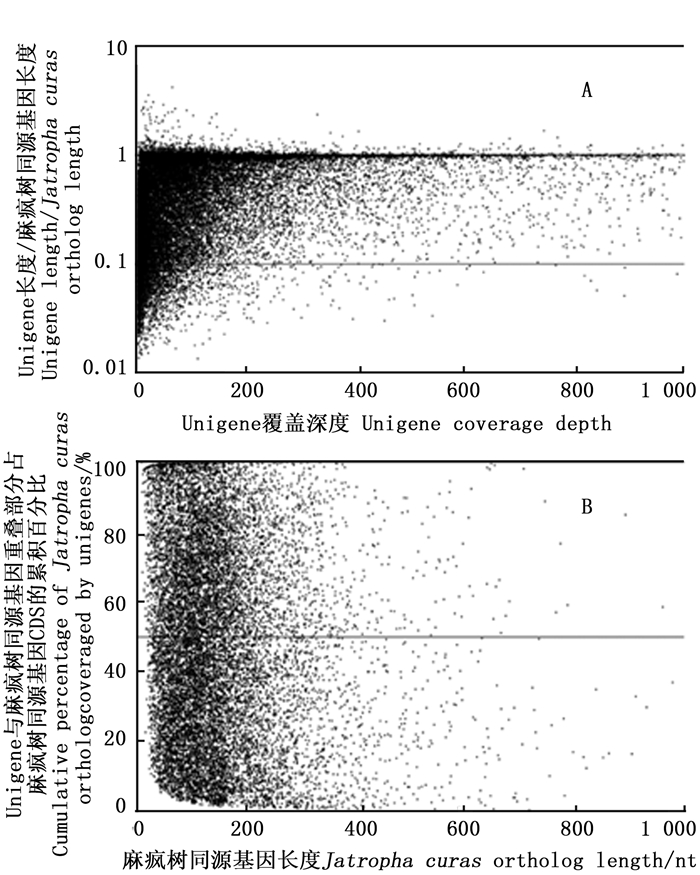
对余甘子Unigenes的CDS长度与近缘模式生物麻疯树同源基因CDS长度的比较分析可知(图 6A):余甘子45 011条Unigenes的CDS中有一半以上(62.96%)可以映射到麻疯树基因组编码区,但仅有25.22% Unigenes的CDS与麻疯树同源基因CDS长度之比接近1(0.9≤Ratio < 1.1);覆盖深度≤300时,20.89% Unigenes的编码区覆盖接近1(0.9≤Ratio < 1.1);覆盖深度>300时,仅有4.33% Unigenes的编码区覆盖接近1。进一步分析可知(图 6B):有近1/6 Unigenes的CDS与近缘物种麻疯树同源基因CDS的覆盖度≥80%,仍有大部分未能覆盖到基因的完整编码区。

图 6 余甘子Unigenes的CDS与近缘物种麻疯树同源基因CDS的比较分布图
Figure 6. Comparison distribution of CDS between Unigenes of P. emblica and orthologous genes of Jatropha curcas
-
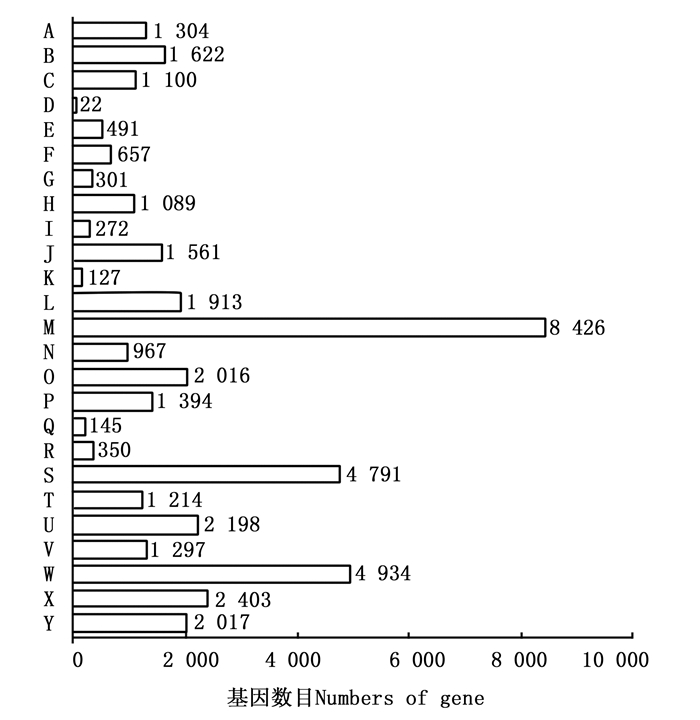
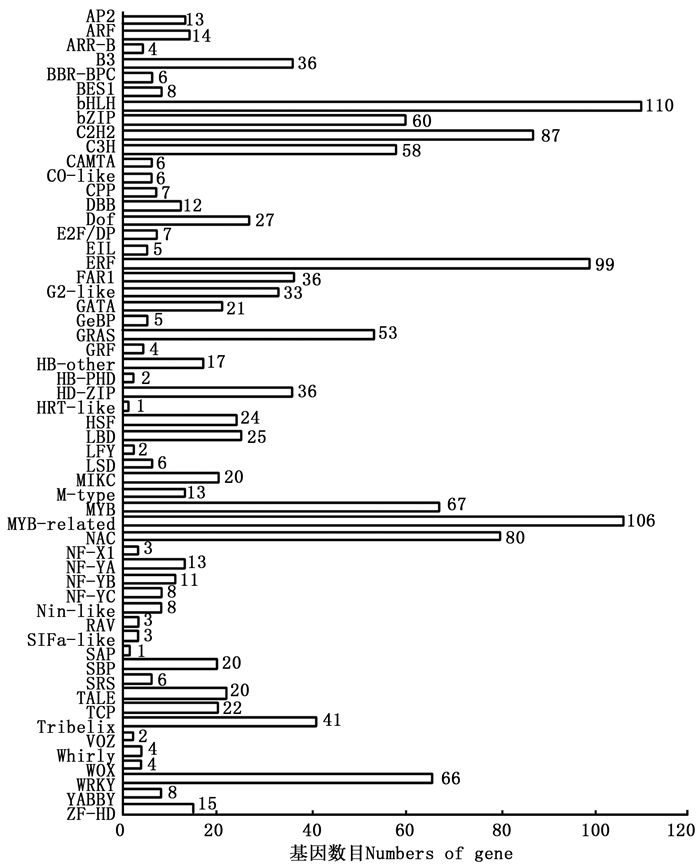
通过PlantTFDB数据库共预测出56个TF家族共1 374个TF的Unigenes(图 7),其中,bHLH、MYB-related、ERF、C2H2、NAC以及MYB类TF为多基因家族,分别占总预测量的8.01%、7.72%、7.21%、6.33%、5.82%和4.88%,而SAP和HRT-like所占比例较少,均为0.073%。对这些TF家族特征进行统计分析可知:共有203条参与植物非生物胁迫应答反应的TF,包括植物中特有的ERF(99条)、NAC(80条)和HSF(24条);WRKY超家族(66条)和bZIP类TF(60条)则与植物生物胁迫的抗性密切相关。这些TF的发现表明余甘子在生长、发育以及适应外界环境过程中基因转录调控的复杂性,也为后续研究余甘子抵抗高温、干旱、盐碱以及病虫害等非生物、生物胁迫提供了新思路。
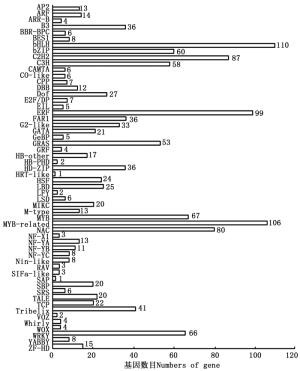
图 7 余甘子转录组Unigenes的转录因子家族分类图
Figure 7. Transcription Factor family classification of Unigenes of transcriptome in P. emblica
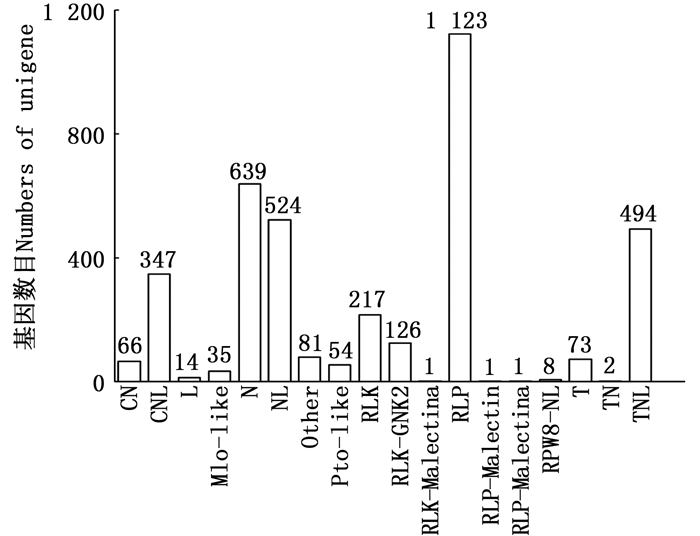
抗性基因(Resistance gene,R-Gene)分布在植物基因组中,其产物介导植物对特定病原体的抗性。本研究预测出18种共3 806条R-Gene的Unigenes(图 8),其中, RLP、N、NL及TNL较多,分别占总预测量的29.51%、16.79%、13.77%和12.98%。这些R-Gene中大多数属于编码包含核苷酸结合位点(nucleotide-binding site,NBS)和/或亮氨酸重复结构(leucine-rich repeat,LRR)保守基序的抗病蛋白,如N基因、RLP基因以及RLK系列基因等[12-13]。

图 8 余甘子转录组Unigenes的R-Gene分类图
Figure 8. R-Gene classification of Unigenes of transcriptome in P. emblica
-
第二代高通量转录组测序技术(RNA-seq)因测序用时短、成本低、数据量大而常被用于转录组学研究中。测序所获得的转录组数据不但能扩充某一物种的基因信息库,在揭示其特定代谢途径的分子机理中也具有重要作用。本研究通过Illumina HiSeq 4000平台对余甘子叶片进行转录组测序,共产生10.95 Gb的数据量,与苦味叶下珠(Phyllanthus amarus Schum. & Thonn.)叶片转录组相比[14],本研究获得的数据信息量较大。一般认为, 当Q30值在80%以上时测序质量就很可靠[15],余甘子叶片转录组Clean reads碱基Q20和Q30分别为98.47%和95.28%,测序质量和准确度比较高。经de novo组装获得76 881条Unigenes,组装的N50值较大,为1 257 nt,组装质量较好;所得Unigenes中长片段较多,且多数集中在200~2 000 nt,近1/4的Unigenes长度在1 000 nt以上,组装序列比较长,为进一步的功能注释、分类、CDS预测和R-Gene预测提供了较好的基础数据。
利用NR、COG、KEGG和SwissProt四大蛋白数据库,将获得的76 881条余甘子Unigenes序列进行比对分析,共有44 768条获得注释,占Unigenes总数的58.23%,而且近一半Unigenes总体表达量FPKM值小于1,表明本研究检测到余甘子低丰度表达的基因比例较高。四大数据库获得注释最多的NR数据库的Unigenes中,14.09%的Unigenes注释为与麻疯树同源的基因,比例高于其它被注释的544个物种,可能是由于麻疯树已完成基因组测序且与余甘子在进化上亲缘关系较近的缘故[16];而仅有33(0.074%)条Unigenes比对到8个同科物种(分属5个属)中,其中,与叶下珠属植物比对上的Unigenes最多,有23(0.052%)条,这可能是现有叶下珠科植物基因组信息较为缺乏或是所得Unigenes多为余甘子特有的新基因而未能比对上相应的基因序列。总的未被注释的Unigenes有32 113条,占总体的41.77%,这些序列可能是因为片段较短,或本身为非编码序列,也有可能是由于这四大蛋白数据库相关数据信息缺乏,或余甘子中存在新基因而未能比对到同源序列[17-19]。
通过COG数据库进行余甘子Unigenes直系同源基因功能注释及分类,获得了大量余甘子在一定时期表达的基因信息,27 008条Unigenes共42 611个功能注释信息涉及到25类基因功能,包含余甘子大部分的生命活动,其中一般功能、信号转导机制、翻译后修饰、蛋白质折叠和分子伴侣的基因表达水平较高,而4.5%(1 913条)的Unigenes生物学功能未知,可能是由于注释信息不足,在很多树种转录组分析中也出现了类似的情况,如长梗杜鹃(Rhododendron longipedicellatum Lei Cai & Y.P. Ma)[18]、云南松(Pinus yunnanensis Franch.)[19]、锥栗(Castanea henryi (Skan) Rehd. et Wils.)[20]等。使用GO基因功能分类体系,将余甘子Unigenes参与的生物学过程、构成的细胞组分和执行的分子功能划分为47个功能亚类,以代谢过程、催化活性和细胞过程的基因表达丰度较高,表明余甘子在这一时期代谢能力较强。利用KEGG进一步研究余甘子Unigenes在生物学上的复杂行为,分析基因产物在细胞中可能的代谢途径,18 175条Unigenes注释到6大代谢通路共21条代谢途径中,其中映射到代谢相关通路的基因数占多数,约为总注释量的3/5,进一步说明余甘子在此时期具有较强的代谢活动。在余甘子KEGG代谢通路中还发现203条与人类疾病相关的Unigenes,包括内分泌和代谢疾病(201条)以及抗药性(2条)。另外,还涉及到氨基酸代谢、碳水化合物代谢、脂类物质代谢、次生物质合成及环境适应等代谢途径,这些数据为探索余甘子抗性机理、挖掘余甘子特殊代谢途径相关基因以及与环境适应性相关基因等研究提供了基础数据。从总体的功能注释信息来看,余甘子基因含量丰富,在分子层面上解释了其具有较强适应性的可能原因。
TF及其结构的多样性决定了其所介导的基因表达调控网络的多样性和复杂性,本研究预测出56个TF家族共1 347个编码TF的Unigenes,这些TF在余甘子生长发育过程中发挥着重要的作用。与拟南芥、棉花、番茄以及杜鹃花等大多数植物不同[18, 21],在余甘子中,数量最多的TF不是MYB家族而是bHLH家族,bHLH家族是植物第二大类TF,可能与余甘子的各种信号转导、合成代谢以及抗逆性等有关[22]。植物体免疫系统能够对抗诸如细菌、真菌、病毒、线虫和昆虫等各类病原体[23],本研究预测到3 806条编码R-Gene的Unigenes,其中,NB-LRR蛋白由植物中最大且最重要的基因家族之一编码,这些NBS-LRR蛋白可以直接或间接地识别病原体分泌的效应物,进而激活下游的信号通路,从而激活植物防御反应,对抗各种类型的病原体[13, 24-25],余甘子R-Gene的预测及分析对了解植物抗病蛋白在植物抗病信号转导途径中的作用机制具有重要的生物学意义,并为余甘子抗病育种提供新的理论指导。
-
本研究使用Illumina HiSeq 4000测序平台构建了余甘子转录组数据库,获得的76 881条Unigenes与四大公共数据库进行比对,有58.23%的Unigenes获得功能注释信息,同时对其进行了CDS预测、TF编码能力预测以及R-Gene预测等分析。研究结果揭示了余甘子丰富的基因信息,在转录组水平上支持了余甘子适应性强的特点,COG、GO、KEGG的功能注释结果均表明余甘子在这一时期进行着较强的代谢活动。所得转录组信息丰富了余甘子基因信息库,为今后余甘子乃至同属物种微卫星标记开发、功能基因挖掘、抗性机理分析、遗传资源分类与进化以及分子辅助育种等研究奠定了重要基础。
云南干热河谷地区余甘子转录组分析
Transcriptome Analysis for Phyllanthus emblica Distributed in Dry-hot Valleys in Yunnan, China
-
摘要:
目的 对云南干热河谷地区余甘子转录组特征进行描述,旨在为余甘子微卫星标记的开发和功能基因的挖掘提供较全面的背景信息。 方法 采用Illumina Hiseq 4000测序平台对余甘子叶片进行转录组测序,对原始数据进行过滤、de novo组装及聚类去冗余等处理后,再与公共数据库进行比对,对Unigenes进行基本功能注释、CDS预测、TF编码能力预测及R-Gene预测等分析。 结果 本研究共获得10.52 Gb的Clean reads,Q20、Q30分别为98.47%、95.28%。组装并去冗余后获得76 881条Unigenes,平均长度、N50分别为713、1 257 nt。通过与NR、COG、KEGG和SwissProt数据库进行比对,44 768条Unigenes获得功能注释。余甘子转录组Unigenes根据COG功能注释信息大致分为25类;按GO功能注释信息划分为生物学过程、细胞组分和分子功能3大类47亚类;参考KEGG注释信息,可归为6大代谢通路、21类代谢途径,其中约3/5为代谢相关通路。根据以上注释结果共检测出42 953个CDS,其余未比对上的Unigenes用ESTScan预测后得到2 058个CDS。同时,预测到56个TF家族以及18种R-Gene。 结论 本研究获得的余甘子转录组Unigenes序列的组装质量较高、完整性较好、基因丰富、功能多样,极大地扩充了余甘子基因信息库,为今后余甘子乃至叶下珠属植物功能基因挖掘、抗性机理分析、分子标记开发、分子辅助育种等研究提供了重要的基础数据。 Abstract:Objective To provide comprehensive genetic information for the development of microsatellite markers and the mining of functional genes in Phyllanthus emblica by characterizing the transcriptome of P. emblica in dry-hot valleys in Yunnan. Method Transcriptome sequencing was conducted on young leaves of Ph. emblica using Illumina Hiseq 4000, followed by filtering, de novo assembly and clustering. Sequence similarity analysis and annotation of the obtained Unigenes were performed based on databases like NCBI-non-redundant (NR) protein database, Gene Ontology (GO), Clusters of Orthologous Groups (COG), KEGG database, SwissProt, PlantTFDB, and PRGdb. Result In total, 10.52 Gb Clean reads with Q20 of 98.47% and Q30 of 95.28% were generated. A total of 76 881 Unigenes with an average length of 713 nt and N50 of 1 257 nt were obtained by de novo assembly and clustering with Clean reads. Out of them, 44 768 Unigenes were functionally annotated against four protein databases. The Unigenes were roughly divided into 25 categories according to COG function, and were grouped into three functional categories (including biological processes, cellular components and molecular function) and 47 sub-categories based on GO functional annotation. KEGG analysis showed that the Unigenes could be fallen into six categories and 21 metabolic pathways, of which about 3/5 were Metabolism. A total of 42 953 CDS were detected based on the results of functional annotation, and 2 058 CDS were predicted using ESTScan with the remaining Unigenes. And 56 Transcription Factor families and 18 resistance genes were predicted. Conclusion The Unigenes of transcriptome in Ph. emblica show high quality, good integrality, abundant genes and various functions, which could lay an important foundation for further study of functional gene excavation, resistance mechanism analysis, molecular marker development and molecular assisted breeding of Ph. emblica and other congeneric species. -
Key words:
- Phyllanthus emblica
- / transcriptome
- / unigene
- / functional annotation
- / CDS
- / transcription factor
- / R-Gene
-
图 1 余甘子转录本和Unigenes的长度分布图
Figure 1. Transcripts and Unigenes length distribution for P. emblica
图 2 余甘子转录组Unigenes的NR注释物种分布
Figure 2. NR annotated species distribution of Unigenes of transcriptome in P. emblica
图 3 余甘子转录组Unigenes的COG功能注释分布统计图
Figure 3. COG functional annotation distribution of Unigenes of transcriptome in P. emblica
图 4 余甘子转录组Unigenes的GO功能分类统计图
Figure 4. GO functional classification of Unigenes of transcriptome in P. emblica
图 5 余甘子转录组Unigenes的CDS长度分布图
Figure 5. CDS length distribution of Unigenes of transcriptome in P. emblica
图 6 余甘子Unigenes的CDS与近缘物种麻疯树同源基因CDS的比较分布图
Figure 6. Comparison distribution of CDS between Unigenes of P. emblica and orthologous genes of Jatropha curcas
图 7 余甘子转录组Unigenes的转录因子家族分类图
Figure 7. Transcription Factor family classification of Unigenes of transcriptome in P. emblica
-
[1] 金振洲, 欧晓昆.元江、怒江、金沙江、澜沧江干热河谷植被[M].昆明:云南大学出版社, 云南科技出版社, 2000. [2] Zhou Z, Ma H, Lin K, et al. RNA-seq reveals complicated transcriptomic responses to drought stress in a nonmodel tropic plant, Bombax ceiba L.[J]. Evolutionary Bioinformatics, 2015, 11(S1):27-37. [3] 李巧明, 赵建立.云南干热河谷地区余甘子居群的遗传多样性研究[J].生物多样性, 2007, 15(1): 84-91. doi: 10.3321/j.issn:1005-0094.2007.01.009 [4] Variya B C, Bakrania A K, Patel S S. Emblica officinalis (Amla) :A review for its phytochemistry, ethnomedicinal uses and medicinal potentials with respect to molecular mechanisms[J]. Pharmacological Research, 2016, 111: 180-200. doi: 10.1016/j.phrs.2016.06.013 [5] Chaphalkar R, Apte K G, Talekar Y, et al. Antioxidants of Phyllanthus emblica L. bark extract provide hepatoprotection against ethanol-induced hepatic damage: a comparison with Silymarin[J]. Oxidative Medicine and Cellular Longevity, 2017, 2017: 1-10. [6] Srinivasan M. Vitamin C in plants: Indian Gooseberry (Phyllanthus emblica)[J]. Nature, 1944, 153(3892): 684. doi: 10.1038/153684c0 [7] Alvarez M, Schrey A W, Richards C L. Ten years of transcriptomics in wild populations: what have we learned about their ecology and evolution?[J]. Molecular Ecology, 2015, 24(4): 710-725. doi: 10.1111/mec.13055 [8] Kumar A, Singh K. Isolation of high quality RNA from Phyllanthus emblica and its evaluation by downstream applications[J]. Molecular Biotechnology, 2012, 52(3): 269-275. doi: 10.1007/s12033-011-9492-5 [9] Grabherr M G, Haas B J, Yassour M, et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome[J]. Nature Biotechnology, 2011, 29(7): 644-652. doi: 10.1038/nbt.1883 [10] Fu L M, Niu B F, Zhu Z W, et al. CD-HIT: accelerated for clustering the next-generation sequencing data[J]. Bioinformatics, 2012, 28(23): 3150-3152. doi: 10.1093/bioinformatics/bts565 [11] Iseli C, Jongeneel C V, Bucher P. ESTScan: a program for detecting, evaluating, and reconstructing potential coding regions in EST sequences[C]// Proceedings of the Seventh International Conference on Intelligent Systems for Molecular Biology. Menlo Park: AAAI Press, 1999: 138-158. [12] 房卫平, 谢德意, 李志芳, 等. NBS-LRR类抗病蛋白介导的植物抗病应答分子机制[J].分子植物育种, 2015, 13(2): 469-474. [13] Chisholm S T, Coaker G, Day B, et al. Host-microbe interactions: shaping the evolution of the plant immune response[J]. Cell, 2006, 124(4): 803-814. doi: 10.1016/j.cell.2006.02.008 [14] Bose Mazumdar A, Chattopadhyay S. Sequencing, de novo assembly, functional annotation and analysis of Phyllanthus amarus leaf transcriptome using the Illumina platform[J]. Frontiers in Plant Science, 2016, 6(340): 1199. [15] 贾新平, 孙晓波, 邓衍明, 等.鸟巢蕨转录组高通量测序及分析[J].园艺学报, 2014, 41(11): 2329-2341. [16] Sato S, Hirakawa H, Isobe S, et al. Sequence analysis of the genome of an oil-bearing tree, Jatropha curcas L.[J]. DNA Research, 2011, 18(1): 65-76. doi: 10.1093/dnares/dsq030 [17] Bai T D, Xu L A, Xu M, et al. Characterization of masson pine (Pinus massoniana Lamb.) microsatellite DNA by 454 genome shotgun sequencing[J]. Tree Genetics & Genomes, 2014, 10(2): 429-437. [18] 李太强, 刘雄芳, 万友名, 等.基于高通量测序的极小种群野生植物长梗杜鹃的转录组分析[J].植物研究, 2017, 37(6): 825-834. [19] 蔡年辉, 邓丽丽, 许玉兰, 等.基于高通量测序的云南松转录组分析[J].植物研究, 2016, 36(1): 75-83. [20] 张琳, 范晓明, 林青, 等.锥栗种仁转录组及淀粉和蔗糖代谢相关酶基因的表达分析[J].植物遗传资源学报, 2015, 16(3): 603-611. [21] 牛义岭, 姜秀明, 许向阳.植物转录因子MYB基因家族的研究进展[J].分子植物育种, 2016, 14(8): 2050-2059. [22] 王翠, 兰海燕.植物bHLH转录因子在非生物胁迫中的功能研究进展[J].生命科学研究, 2016, 20(4): 358-364. [23] Jones J D G, Dangl J L. The plant immune system[J]. Nature, 2006, 444: 323-329. doi: 10.1038/nature05286 [24] Gururani M A, Venkatesh J, Upadhyaya C P, et al. Plant disease resistance genes: current status and future directions[J]. Physiological and Molecular Plant Pathology, 2012, 78(51): 51-65. [25] Dubey N, Singh K. Role of NBS-LRR proteins in plant defense[M]// Singh A, Singh I. Molecular Aspects of Plant-Pathogen Interaction. Singapore: Springer Singapore, 2018: 115-138. -
 点击查看大图
点击查看大图

计量
- 文章访问数: 5042
- HTML全文浏览量: 1735
- PDF下载量: 558
- 被引次数: 0
